
Newest releases from Nucleus and Herasight
Nucleus releases sibling validation paper, Herasight releases imputation validation paper

Things are moving fast in the embryo selection sector. There was never really any technological barriers here the last 10+ years, just the usual social taboos and various incompetent people in the sphere. While we wait for the taboos to die, new players have sidelined the older companies (mostly Genomic Prediction since the other companies apparently gave up). Anyway, there are again news from Nucleus and Herasight, so let’s go over them in turn.
Nucleus genomics validation paper
Nucleus posted their validation study on a preprint server on Oct 28th, presumably in reply to Herasight’s. But I only had time to read it in full now, so I will do so. I also want to say kudos to them for sending me their scoring files which I discussed in my last piece over at Aporia. These are the files they used for scoring in this study.
Cordogan, S., Starr, D. B., Treff, N. R., Lanchbury, J., Goldstein, E., Sadeghi, K., ... & Folkersen, L. (2025). Within-and Between-Family Validation of Nine Polygenic Risk Scores Developed in 1.5 Million Individuals: Implications for IVF, Embryo Selection, and Reduction in Lifetime Disease Risk. medRxiv, 2025-10.
Polygenic risk scores (PRSs) can reduce lifetime disease risk by guiding embryo selection during in vitro fertilization (IVF). We performed genome-wide association meta-analyses totaling ∼1.5 million individuals to construct state-of-the-art PRSs for nine diseases: Alzheimer’s disease, breast cancer, coronary artery disease, endometriosis, hypertension, prostate cancer, rheumatoid arthritis, type 1 diabetes, and type 2 diabetes. The resulting predictors achieved liability-scale R2 values of up to 22.9% for type 2 diabetes, matching or exceeding previously published benchmarks across all scores. Three PRS - Alzheimer’s disease, prostate cancer, and type 2 diabetes - explained over 75% of the common-SNP heritability. Within-family validation in 40,872 siblings across 18,840 families showed that, for eight of nine diseases, predictive performance was comparable to population-level results, confirming substantial direct genetic effects. Modeling of embryo selection suggests that couples with five euploid embryos could achieve 27-67% relative risk reduction across diseases. While the limitations of genetic data availability meant that these estimates were performed in European ancestry samples, we performed validation in the multi-ancestry US-based All of Us Biobank, demonstrating significant statistical power across ancestries. These findings support the clinical applicability of PRS-guided embryo selection to reduce the burden of common diseases.
Fun fact is that it includes Nathan Treff on the author list. He was a cofounder of Genomic Prediction back in 2017. Since jumping ship, the old failing company has decided to sue him for clown points. Their claims are:
Nucleus Genomics and two former Genomic Prediction executives were hit with a trade secret misappropriation lawsuit on Oct. 22 in New Jersey District Court. The complaint, filed by Troutman Pepper Locke on behalf of DNA testing company Genomic Prediction, accuses former chief science officer Nathan Treff and former head of medical affairs Talia Metzgar of stealing confidential information before joining Nucleus, which had previously attempted to acquire Genomic Prediction. According to the suit, Treff deleted data from his company laptop and disabled security cameras before his abrupt resignation in August 2025, while Metzgar emailed 30 confidential documents to her personal account, enabling Nucleus to rapidly expand into embryonic DNA testing using Genomic Prediction’s proprietary methods. The case is 2:25-cv-16850, Genomic Prediction, Inc. v. Treff.
Sounds like normal startup company behavior. Presumably, Nucleus will pay for any settlements. Anyway, let’s move on to their validation paper. It concerns the 9 diseases they built prediction models for. They were able to pool some great datasets: “Million Veterans Program (MVP), Finngen, the UK Biobank (UKBB), All of Us (AoU), and various trait-specific consortia”. They are missing some cohorts, e.g. the Japanese Biobank. Their comparison figure is:
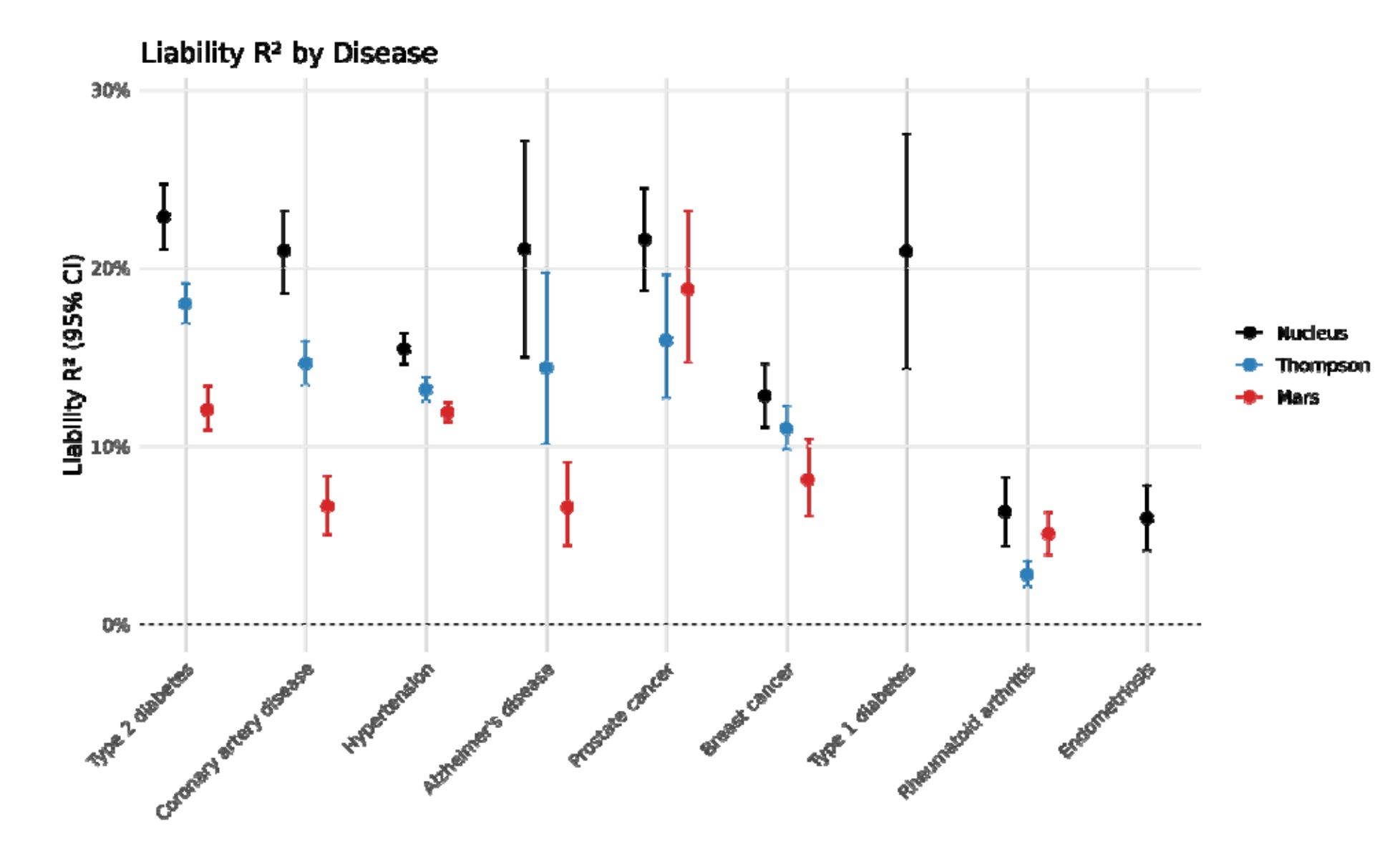
It’s a pretty low quality figure in the source unfortunately. Their comparisons are some academics. It would have been sensible to compare with the other embryo selection companies in addition. Just like Herasight, they used the UKBB sibling comparisons:
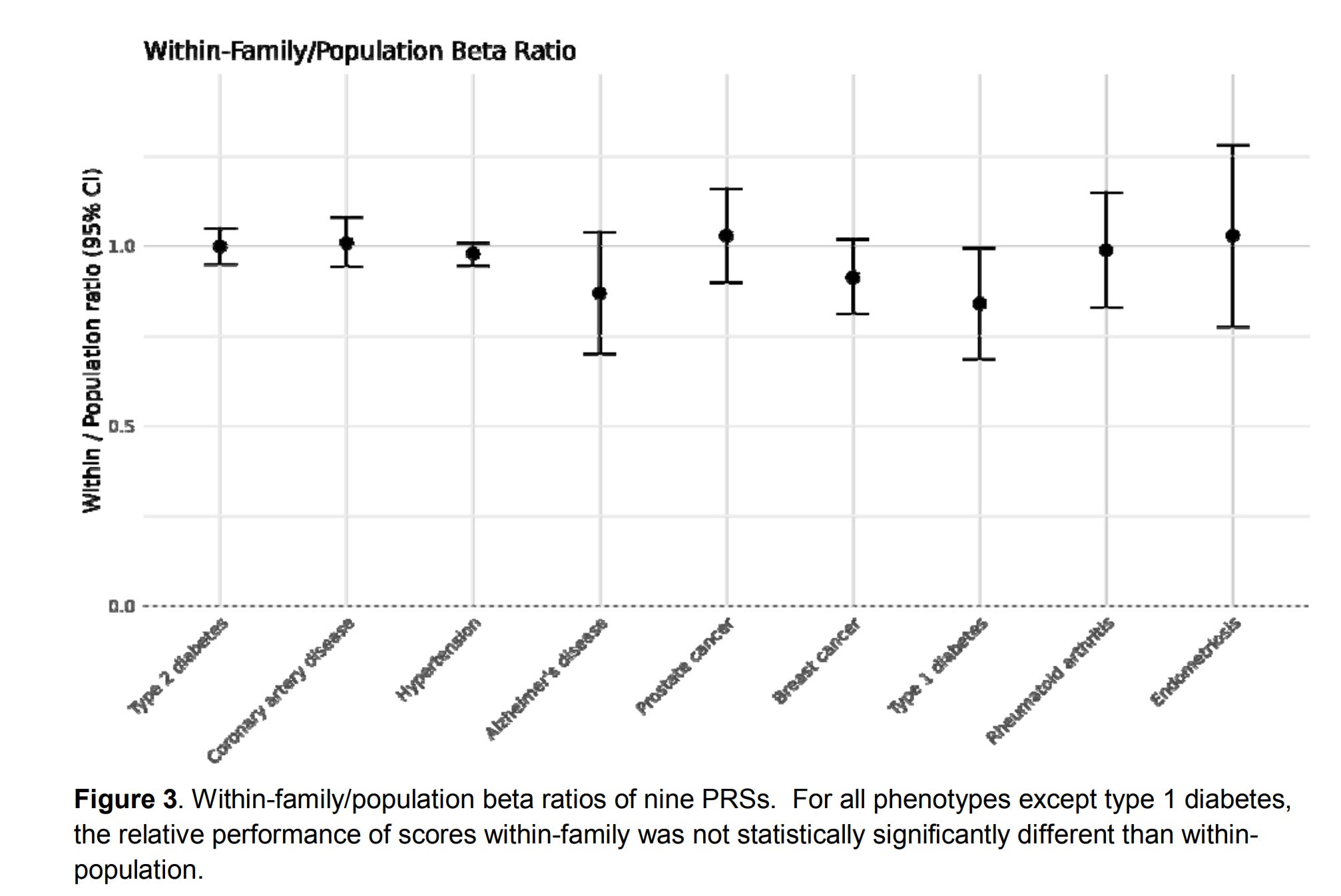
They aren’t terribly clear how this was done, simply saying:
We adapted methods for within-family PRS validation from Selzam et al. for binary phenotypes (21), and calculated the ratio of within-family to population performance. We found that the within-family predictive power of the PRSs was not significantly different from the population predictive power for eight out of nine scores, with the exception of type 1 diabetes. The Liability R2 of type 1 diabetes was reduced accordingly for risk reduction calculations in the context of embryonic selection.
But the ratio of what? The liability incremental R²? The betas? Ratios of variances is a bad idea (variances in general are a bad idea for human use).
Anyway, using their validities they also compute the disease reduction (relative rate reduction) from choosing the best of X embryos using these models. The results look pretty normal, say, 77% lower risk of type 1 diabetes if picking lowest risk among 10 (and other counts of) embryos. These kind of result should not be over-interpreted. For instance, their results show a 67% reduced chance of Alzheimer’s. However, if you live long enough, you always get Alzheimer’s (and everything else), so in some sense we could say that everybody and every embryo has 100% chance of every disease. The numbers here are only relevant to the comparison group, which is some old British people in the UK biobank. At some given age, only some subset of people will get a given disease, and this chance can be statistically reduced by having different genetic variants. As such, what the number says is that hypothetically, if all the embryos were grown and implanted, then the lowest predicted risk one would have -2/3 chance of getting Alzheimer’s by whatever age the UKBB sibling sample has (50-70s). The same applies to all the other predictions and estimated ‘treatment’ effects.
Anyway, it is a bit strange they didn’t cite their competitor’s work from August (cf. preprint, blog, CTRL+F finds no “Moore” et al in their study), since presumably they had 2 months to make comparisons and reply to criticism. What the potential customer wants is a direct comparison so they know who to go with. Sadly, this validation does not provide that and is also silent against the criticism Herasight made of their prior results. Maybe they are saving it for their next paper. This paper is essentially a copy of the Herasight paper as it is.
Herasight imputation methods paper
Herasight’s new paper concerns their imputation methods and as before features a companion blogpost:
Li et al. 2025. ImputePGTA: accurate embryo genotyping and polygenic scoring from ultra-low-pass sequencing.
Preimplantation genetic testing for polygenic risk (PGT-P) holds great promise for reducing lifetime disease burden, but has been held back by the difficulty of genotyping embryos. Preimplantation genetic testing for aneuploidy (PGT-A) is a standard-of-care technology used in over half of in vitro fertilization (IVF) cycles in the United States. PGT-A is used to detect chromosomal abnormalities using ultra-low-pass (ULP) sequencing data (typically 0.002x to 0.006x) or, less commonly, genotyping array-based data. Here we describe ImputePGTA, a Hidden Markov Model-based algorithm that enables accurate reconstruction of embryo genomes from array or ULP sequencing data from embryos and parental genome data. A key innovation of our algorithm is its ability to provide accurate embryo genotypes and polygenic scores (PGSs) along with posterior distributions given limited embryo data and imperfectly phased parental haplotypes, as encountered in real-world applications. The accuracy of the embryo genome reconstruction increases with that of the phasing quality of parental haplotypes. We describe a method, phaseGrafter, that improves parental phasing by combining statistical phasing from short-reads with read-backed phasing from long-reads, which further enable phasing of rare pathogenic variants. We validate our results through simulations, downsampled gold standard data, and comparison of six reconstructed embryo genomes from real PGT-A data to high-coverage, post-birth whole genome sequencing data. Our imputed embryo genotypes have a dosage correlation of 0.961 with high-quality post-birth genotypes (0.998 when using embryo array data). The imputed embryo polygenic scores for 17 diseases have a mean absolute difference of 0.16 standard deviations (0.023 when using embryo array data) with PGSs calculated from high-quality post-birth genotypes, lower than from imputation of array data from reference panels. We show that the attenuation in expected gains from embryo selection due to posterior uncertainty is only ~5-10% for typical PGT-A data. Our approach removes an important technological barrier to using PGT-P and will facilitate more widespread adoption.
The business problem is this: there are 1000s of IVF clinics. Many of these provide some kind of genetic testing. This genetic testing is done using older systems, like miSeq 500, but generally is random reads very low coverage. In other words, the DNA from the embryo is copied a large number of times (using PCR), and then broken into smaller chunks. Some of these chunks are then read at random. Typically, a given basepair (like AT) is then read on average 0.005 times (coverage = % of genome read at least 1 times; depth = average reads per variant in genome; these values are approx. the same at the low end since variants are read either not at all, or once, and only rarely more than once). Which is to say that it is not read 99%+ of the time. This poor quality data is enough that one can determine the number of chromosomes (PGT-A for aneuploidy), since one just counts the number of reads mapped (thought to belong to) a given chromosome to the expected number given the total number of reads and the size of the chromosome. So if an embryo has 5k reads on chromosome 5, but the expected count was 10k, then it it seems to have half the expected number of copies, that is, it has only 1 copy of chromosome 5 (instead of 2) and should be discarded (monosomies are always nonviable). Given the low quality data, usually the estimates aren’t terribly precise, but in many cases good enough to make a call (use/don’t use). Additionally, especially if there is some family history, clinics may perform additional targeted sequencing (PGT-M for Mendelian) where they measure particular locations on the genome with high priority genes. Say, BRCA genes for female cancers. These are then reads 100s of times so a very confident call can be made for which variants are present.
The problem with the data from these methods is that one cannot use them for scoring polygenic scores for phenotypes that are spread out in the genome. Since most phenotypes have such a distributed genetic architecture, data from PGT-A and PGT-M methods never concerns most of the relevant variants, and in the rare case that a variant is read once, there is no certainty about it due to inherent errors in the sequencing methods. So what to do? Well, one can impute genetic data using reference panels. We are quite familiar with this method from ancient genomes, and generally just upgraded array data (which typically measures 600k-2M snp-like variants with precision). The problem is that imputation using reference panels typically doesn’t work well when the sequencing depth (reads per variant) is much lower than 1x. However, this changes a lot if one has family data. Biologically speaking, the DNA present in a given embryo can only come from 2 places: mom and dad. Thus, if one knows the parental genomes with high precision (like 30x sequencing), all one needs to do is figure out which chunk at a given location came from which parental chromosome:
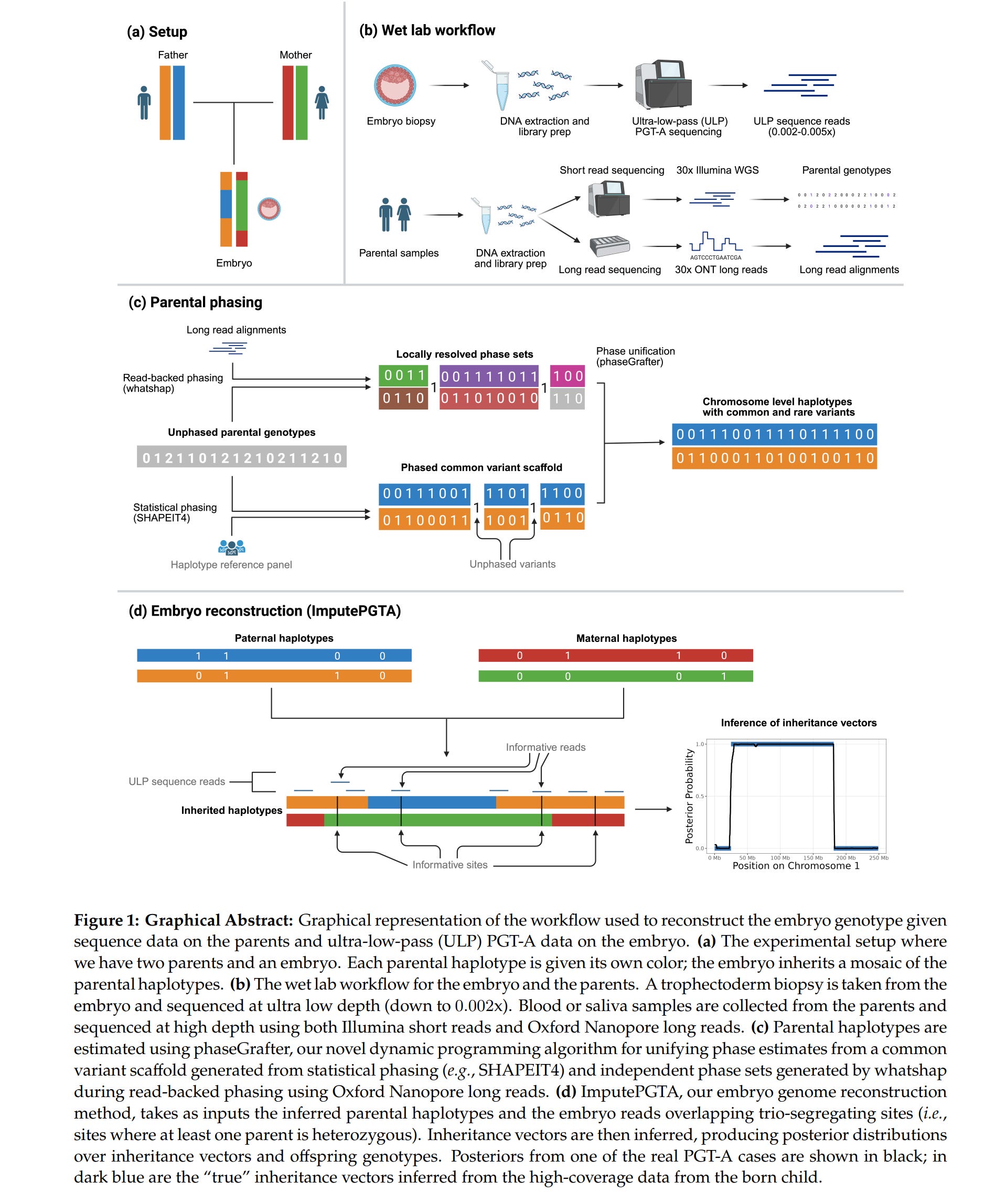
Looking at their helpful illustration we can see that for a given chromosome pair in an embryo, the embryo has received the blue chromosome material in the middle part and the orange chromosome elsewhere, both from dad. From mom, the green chromosome material is in the middle and the red one elsewhere. These transitions are called the breakpoints because it is where recombination occurred. To impute the data from parents, one must figure out where these breakpoints are, and before that, one must figure out which variants are on which parental genome. This process is called phasing. In general, family phasing is done using informative sites. To see how this works, imagine that dad has TT at some location, while mom has AT. Thus, no matter which chromosome embryo got dad’s DNA from, it will get T. However, for mom, it will get A or T depending on which chromosome it came from (red or green). If we see the embryo is AT, we know the DNA in this location came from mom’s chromosome with T (read errors aside). Then one repeats this across the genome for all places where such an informative pair is perhaps present and for each parent. This will give one a scattered map of parental origins of the DNA. Then one does some error correction and essentially fills in the gaps. And voilá, one has in theory phased the data to the parental genomes, assuming assumptions. An alternative simpler and stronger approach is to take a single cell and physically separate the chromosomes for individual sequencing which would give one ~100% confidence in the phasing. It seems this method has been around for a while and here’s a 2018 paper about it. Anyway, they validated their statistical and biological approach on external data:
We benchmark the model in silico using the Platinum Pedigree 43, a publicly available gold-standard dataset, by both simulating offspring with PGT-A data and downsampling reads from the real offspring data. We perform real-world validation of our approach using data from six embryos across four families that underwent PGT-A testing (2 with ULP data and 2 with array data), subsequent implantation, and live birth. We examine the performance of our method for estimation of embryo PGSs for 17 diseases by comparing to PGSs computed from high quality, post-birth genotypes, obtaining a mean-absolute difference of 0.157 SDs for embryos with ULP sequencing data and 0.0298 SDs for embryos with genotyping array data. Our results show that accurate genome reconstruction and polygenic scoring, along with posterior uncertainties, can be achieved from routine PGT-A data, removing an important barrier to wider adoption of PGT-P.
So, using this PGT-A-data imputation and scoring, there is on average only a 0.16 SD difference in the polygenic scores on average. This is a quite small difference, which would only rarely cause one to use or discard the wrong embryo. This is good enough for practice.
The general alternative to this imputation approach is to measure the DNA of embryos using better technologies. There are a variety of options from single cell sequencing, deep sequencing, to petri dish sequencing (some DNA flows out of the embryo). The other companies used these approaches (Genomic Prediction uses arrays, Nucleus uses deep sequencing). These methods are probably better in general, but their problem is that they create huge bottlenecks since clinics aren’t able to do these advanced genotyping methods. Thus, one would have to ship frozen embryo tissue samples to wherever the lab is. The imputation approach only requires that clinics do the same thing they are already doing.
Next up, given the current imputation accuracy, how large is the decline in embryo selection accuracy?
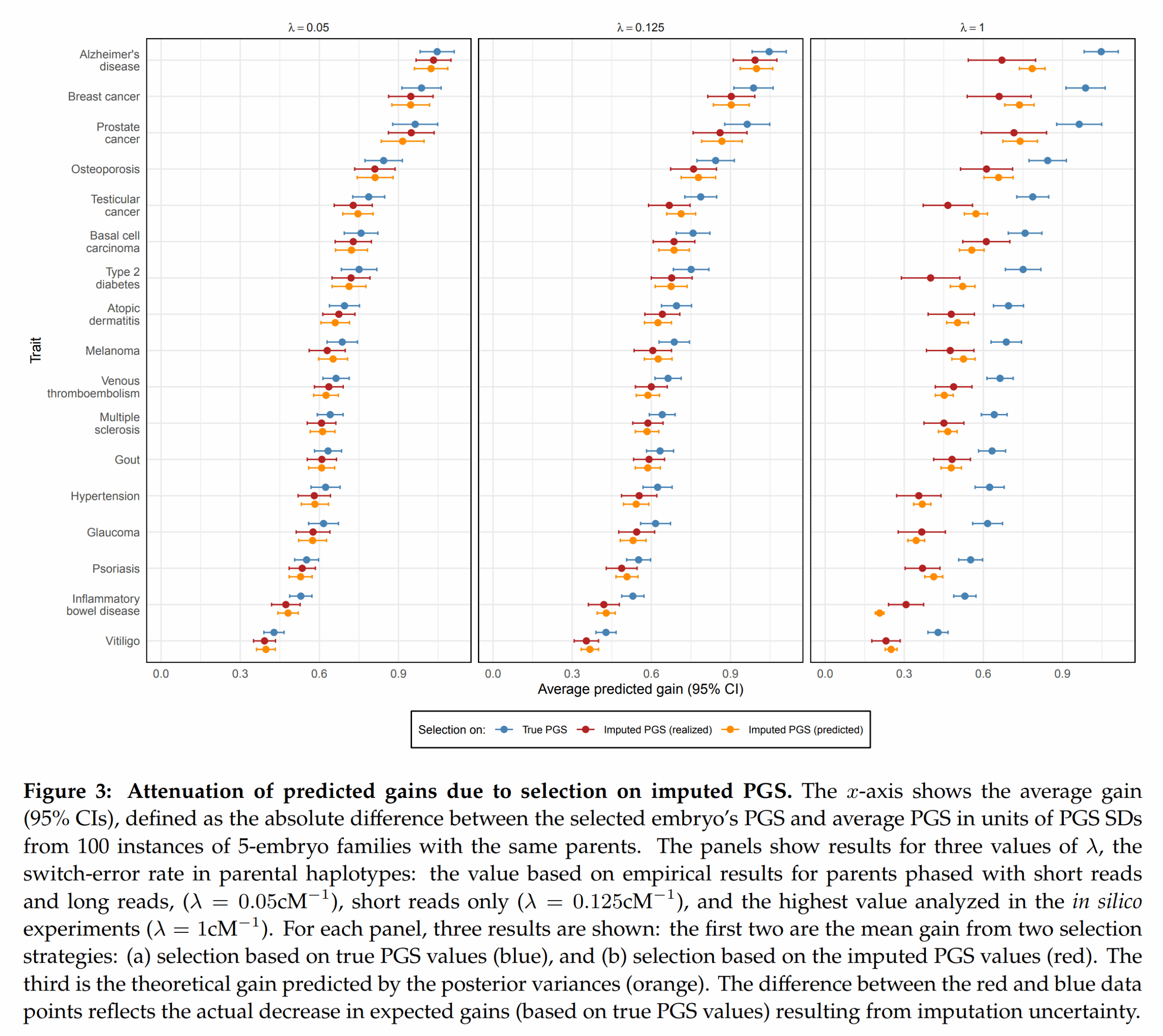
They did some modeling to derive theoretical predictions of how large the decline would be (the usual classical test theory considerations), and then downsampled some real data to see if simulations using real data agreed with theoretical predictions. Fortunately, they mostly did (yellow error bars overlap red means). It looks like when the switch errors are very high, the model fails a bit (right side plots shows yellow and red dots far apart in some cases, like IBD).
But one can go further, and try this on real embryo data too. Some people used Genomic Predictions’s product and others used some standard PGT-A approach for choosing embryos, and then later had children, who could be then sequenced. These gold standard DNA data can then be compared to PGT-A imputed data and scores for the same models:
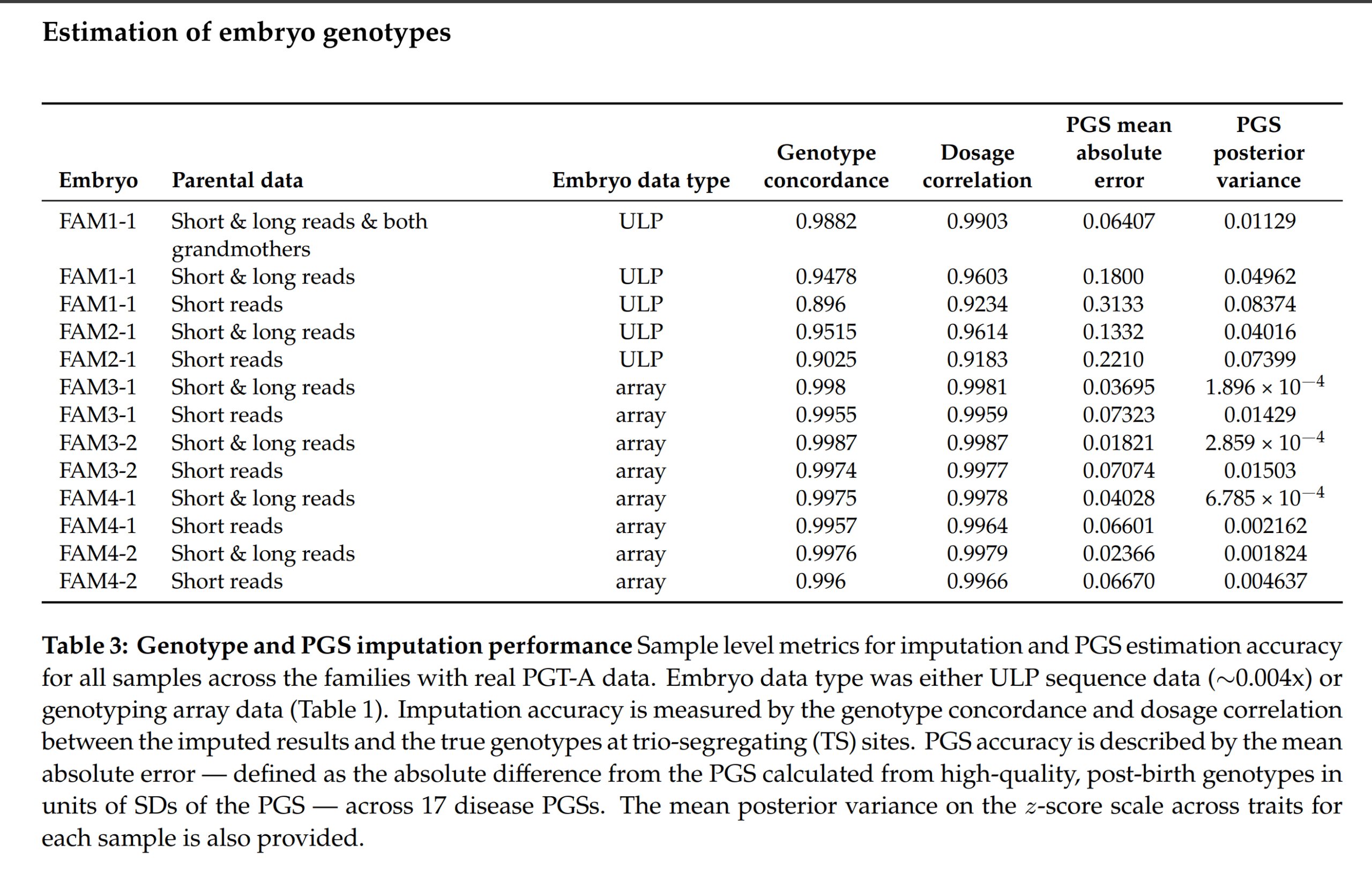
I say some of them used Genomic Prediction’s service because they have array-data for embryos and I think it’s the only way to get this aside from running your own lab. Anyway, for the cases with data from low quality sequencing (ULP), we can see that the real polygenic scores errors are on average around 0.20 SD (vs. 0.16 theoretical prediction). Pretty good!
In fact, one can use more family members to get more accuracy with the imputation. One chief problem is that phasing the parents themselves is also difficult. Unlike what the schematic shows, we can’t really in practice figure out with 100% accuracy which chromosome a given letter is on. Statistical phasing using a bunch of mostly unrelated (very distant cousins AKA same-race) people can only get you so far, maybe to 99% accuracy. If one has data from other family members, however, the accuracy increases. Grandparents are best, but parental siblings and so on also work. They show this to be the case using some of the families which had such family data:
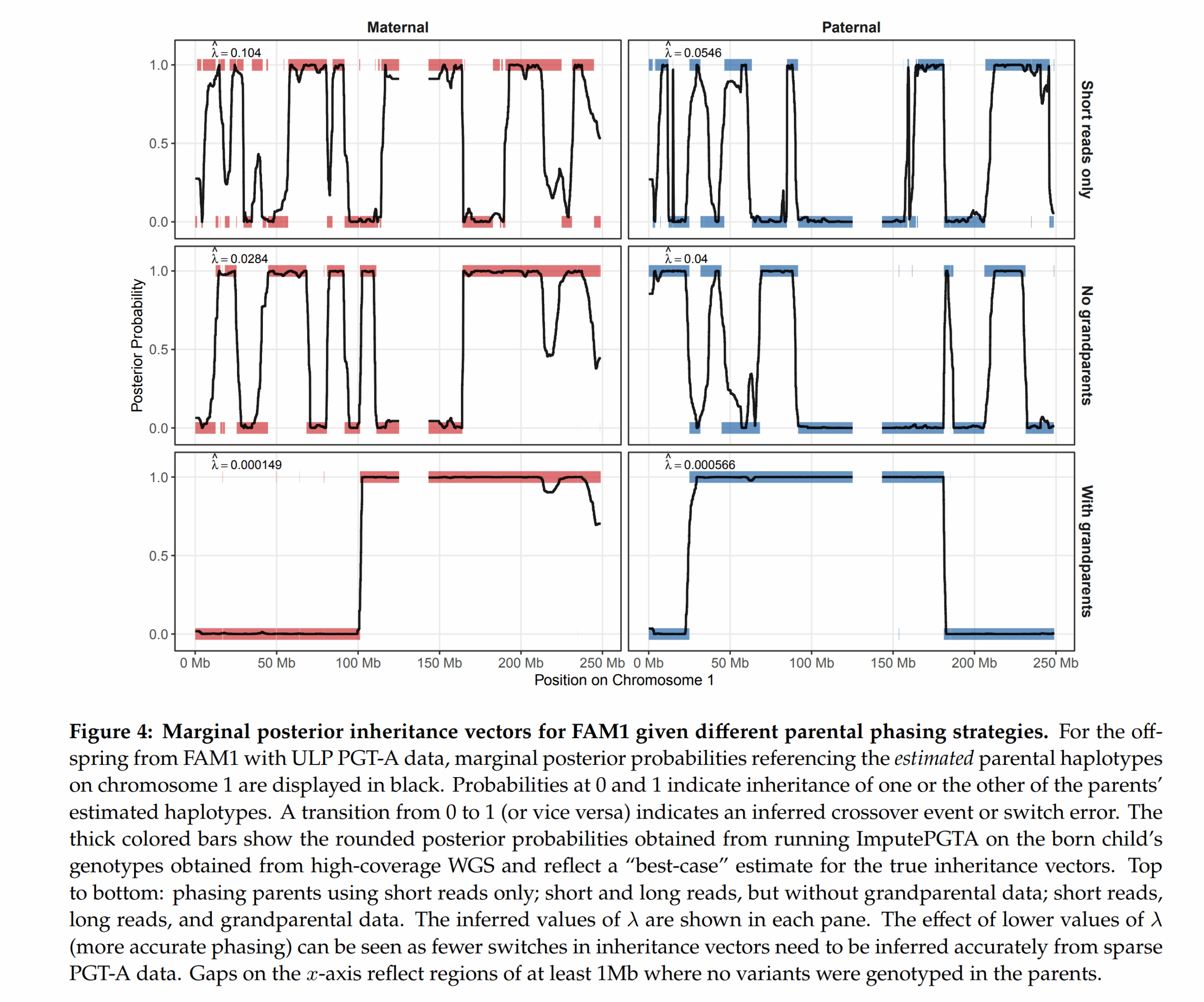
The upshot of this is that embryo selection may be coming to a place near you as well. Their blogpost says they are offering a 90% discount if you are the first customer from a given country. If all you need to do is do IVF at the local clinic and get whatever PGT-A data they offer, then send it to Herasight, things get a lot easier. Of course, beware that many countries ban advanced embryo selection, so you may have to fly to America or some other place. Existing law does not seem clear on exactly where the ‘selection event’ takes place. Herasight and Nucleus are US-based companies, but parents ultimately pick the embryo they want. If the embryos are physically present in the USA, but parents are physically in [your country], and USA sends the embryo to [your country], does the selection take place in USA or [your country]? Politicians shouldn’t make such silly laws to begin with. Who is liable for doing any illegal selections? Parents or everybody involved? They should just get rid of these laws.
It’s exciting to see all the recent progress in embryo selection. Maybe Nucleus and Herasight can have a productive consumer eugenics arms race, like Nvidia vs. ATI (acquired by AMD) for GPUs, or Intel vs. AMD for CPUs. In general, it would be better with more players in the field.
I have the IQ report from nucleus my initial score based on 500k genes was 95th percentile with a predicted IQ of about 120 but now that they updated their model to include 7 million genes my score dropped to only an IQ of 102-103 supposedly these scores shouldn’t be as accurate for me because I’m not northwest European but Sephardic Jewish… I saw on some website that the predictive power of these genetic models is most accurate for northwest Europeans but less accurate for Italians or poles even
One more competitor would be excellent, this space surely still has plenty of... space.
A bit of competitive pressure can do amazing things for progress.